ASOCIACIÓN CÁNTABRA DE FIBROSIS QUÍSTICA
¿Qué es la fibrosis quística?
La Fibrosis quística (FQ) es una enfermedad hereditaria que se debe a un defecto genético que afecta a múltiples sistemas del organismo.
La Fibrosis Quística es una enfermedad crónica y hereditaria que representa un grave problema de salud. Es una enfermedad degenerativa que afecta principalmente a los pulmones y al sistema digestivo.
Consiste en una alteración genética que afecta a las zonas del cuerpo que producen secreciones, dando lugar a un espesamiento y disminución del contenido de agua, sodio y potasio originándose la obstrucción de los canales que transportan esas secreciones y permitiendo que dicho estancamiento produzca infecciones e inflamaciones que destruyen zonas del pulmón, hígado, páncreas y sistema reproductor principalmente. Es una patología grave de tipo evolutivo con una esperanza de vida limitada y que hoy día no tiene curación.
Para mantener controlada la enfermedad, las personas con Fibrosis Quística necesitan un cuidado permanente con continuos tratamientos para las complicaciones pulmonares y digestivas, con controles periódicos en el hospital y una dedicación plena por parte de los pacientes y de su familia.
Un diagnóstico precoz puede prolongar la esperanza de vida de las personas con Fibrosis Quística y mejorar la calidad de la misma. En los últimos años se ha avanzado mucho en el conocimiento y tratamiento de la enfermedad pero, a pesar de eso, sigue siendo una patología sin curación. Cuando la enfermedad se encuentra en un estadio muy avanzado, existe la posibilidad del trasplante pulmonar y/o hepático. Se estima que la incidencia de la Fibrosis Quística en nuestro país es de un caso de cada 3500 nacidos vivos, mientras que uno de cada 30 habitantes son portadores sanos de la enfermedad.

LA FIBROSIS QUÍSTICA
Sintomas
La enfermedad suele manifestarse por problemas respiratorios asociados con manifestaciones digestivas, diarrea crónica y retraso del crecimiento. Esta es la forma de presentación más frecuente. Sin embargo, a lo largo de la vida pueden aparecer signos y síntomas que configuran la historia natural de la enfermedad.
En el recién nacido se observan: retraso en la evacuación del meconio, ictericia prolongada o anemia, hipoproteinemia y edemas. No es infrecuente que se manifieste con ileo meconial y peritonitis meconial. En el lactante las alteraciones respiratorias pueden ser la primera manifestación: tos de tipo “tosferina”, broncoespasmo o bronconeumonías de repetición. No es raro etiquetar a estos niños de asmáticos o alérgicos. Suelen aparecer en este periodo los primeros síntomas de insuficiencia pancreática. Este cuadro va empeorando durante la edad preescolar y escolar, haciendo un cuadro clínico más florido, con múltiples complicaciones fundamentalmente pulmonares, infecciones constantes que destruyen el pulmón conduciendo a la muerte. El trasplante pulmonar puede representar la última opción terapéutica en estas personas.
LA FIBROSIS QUÍSTICA
Diagnóstico
Actualmente la Fibrosis Quística esta incorporada al programa de cribado neonatal , por lo que la enfermedad puede detectarse al nacimiento, extrayendo una pequeña gota de sangre del talón del bebe.
En el caso de no existir cribado neonatal el diagnóstico de FQ se realiza por criterios clínicos y de laboratorio.
Ante un paciente con historia familiar o datos clínicos sospechosos se debe realizar el test del sudor. En caso de prueba positiva el diagnóstico se confirma. En casos dudosos o para completar el estudio se realizan estudios genéticos.
El test del sudor es una prueba que determina la concentración de cloro en el sudor. Existen otras enfermedades que se asocian a un test del sudor alterado, por lo que para considerarlo válido se debe repetir dos o tres veces con resultados coincidentes. Si la prueba es dudosa o incluso normal, y el paciente presenta síntomas compatibles se puede realizar otra prueba que mide el potencial bioeléctrico en el epitelio respiratorio.
Siempre se debe hacer estudio genético ante la sospecha de enfermedad. En muchos casos no se detecta ninguna mutación, pero esto no descarta el diagnóstico. En parejas con hijos afectados se puede realizar diagnóstico prenatal. El estudio de portadores queda restringido a las familias de pacientes con FQ.
LA FIBROSIS QUÍSTICA
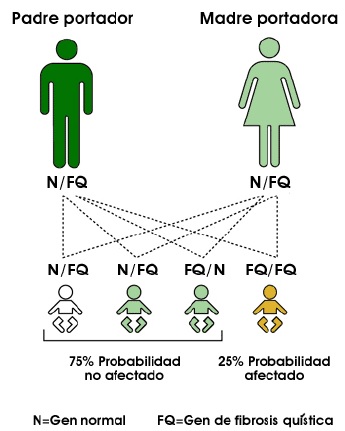
¿Cómo se transmite?
La fibrosis quística es una enfermedad genética hereditaria que se trasmite de forma autosómica recesiva, es decir afecta a ambos sexos, y se necesitan dos progenitores portadores del gen de la Fibrosis Quística, para que exista la posibilidad de tener un hijo con esta enfermedad. La probabilidad de que dos portadores tengan un hijo con Fibrosis Quistica, es de un 25% , la probabilidad de que tengan un hijo sano portador del gen es de 50%, y la probabilidad de tener un hijo sano no portador es del 25%. Estas probabilidades se repiten en cada embarazo.
La Fibrosis Quística es la enfermedad de carácter hereditario más frecuente en la raza blanca y la causa más común de enfermedad pulmonar crónica tanto en la infancia como en la adolescencia.
La incidencia de la enfermedad en Europa oscila entre 1/2.500 y 1/4.500. Mientras que en la raza blanca americana la incidencia es de alrededor de 1/2000, en la raza negra, tanto en americanos como en africanos la incidencia es menor, de aproximadamente 1/17.000. La enfermedad es rara en indios y orientales.
ASOCIACIÓN CÁNTABRA DE FIBROSIS QUÍSTICA
Tratamientos
A continuación mostramos varios tratamientos que pueder resultar beneficiosos.
La afectación pulmonar requiere: en primer lugar fisioterapia respiratoria que es imprescindible para conseguir un drenaje efectivo de las secreciones espesas que presentan los pacientes. Esta debe llevarse a cabo de forma diaria y controlada. Se aconseja la práctica deportiva en función de la capacidad individual de cada persona.
Entre estos protocolos de atención a la persona con FQ, los aspectos nutricionales constituyen una parte muy importante, ya que está demostrado que la malnutrición condiciona un empeoramiento de la función pulmonar y por tanto de la supervivencia. Por ello se ha propuesto al estado nutricional como factor pronóstico, predictor de la morbi-mortalidad en la FQ.
Son múltiples los factores que justifican el riesgo nutricional de estas personas.
La ingesta: en muchas ocasiones el apetito está comprometido en función de:
- Los episodios infectivos.
- La mucofagia.
- El reflujo gastroesofágico.
- Las abdominalgias postpradiales ligadas a maldigestión.
El gasto. El gasto energético GE) está incrementado en estas personas.
- GE basal.
- GE por actividad física.
- GE por termogénesis
Las pérdidas.
En las personas con insuficiencia pancreática (cerca del 85%), la maldigestión condiciona una importante pérdida de energía y nutrientes, La aparición de intolerancia a la glucosa y de diabetes, cuya frecuencia se incrementa con la edad, condiciona también un defecto en el aprovechamiento intracelular de esta fuente energética.
Aporte energético:
se recomienda que la dieta aporte entre un 120-150% de la energía recomendada para la edad y sexo Desde el punto de vista dietético, el aporte suplementario de energía debe realizarse con alimentos naturales, respetando los horarios habituales, pero fragmentando el número de comidas a no menos de 5 diarias. Se elegirán alimentos de alta densidad calórica, para no sobrecargar volumétricamente el estómago. Clásicamente se recomiendan chocolate, fritos, snaks, etc., pero la opción más idónea son los frutos secos.
Los consensos sobre Nutrición en FQ aconsejan que un 15-20% de la energía se aporte como proteínas, un 40-48% como carbohidratos y un 35-40% como grasa. Las personas con FQ muestran un perfil lipídico con deficiencia en ácidos grasos esenciales y especialmente en DHA y EPA (primaria o verosímilmente secundaria a la malabsorción). Este perfil, con mayor tendencia a la síntesis de eicosanoides proinflamatorios, podría ser responsable de una mayor susceptibilidad a infecciones pulmonares. Por este motivo se están realizando investigaciones sobre el efecto de la suplementación de la dieta con distintas concentraciones de ácidos grasos de las series y sobre todo de la ; así como con DHA y EPA para favorecer la síntesis de proteínas con menor capacidad proinflamatoria.
A pesar de los esperanzadores resultados preliminares, aún no hay consenso sobre las recomendaciones de su utilización, dosis, tiempo de administración etc..
De momento sólo se aconseja alto consumo de pescado, aceite de borraja, etc
Los pacientes con riesgo de pérdida salina excesiva por el sudor (fiebre, golpe de calor, ejercicio físico intenso) deben recibir un suplemento de ClNa y en general se aconsejan comidas ligeramente saladas. Los suplementos de Fe, Mg o de Zn dependerán de si existe o no deficiencia. El riesgo de osteoporosis es alto, tanto por la formación de jabones cálcicos con la grasa no absorbida, como por el déficit de vitamina D. También colabora la inactividad ligada a las infecciones repetidas y la hepatopatía. Por ello, cuando la dieta no asegura la ingesta de Ca recomendada debe administrarse éste como suplemento. En todo caso, la vigilancia anual del riesgo de osteoporosis, indicará el tiempo que debe mantenerse esta profilaxis.
- Indicaciones de soporte nutricional
-
- Cuando el % de peso para la talla (Índice de Waterlow I), tanto en niños, como en adultos, es inferior al 85%.
- Índice de masa corporal inferior al percentil 25 en niños y a 18,5 en adultos.
- En niños con estacionamiento ponderal en dos controles sucesivos (2-6 meses).
- En cualquier edad: pérdida ponderal > 5% en dos meses.
- Pacientes normonutridos o con malnutrición leve los que se prevé una alimentación oral insuficiente más de 7 días.
- Riesgo de desnutrición por infección grave y/o tratamiento agresivo, cuya ingesta no cubre el 75% de las recomendaciones
- Suplementar la dieta oral con alimentos naturales
- Debe intentarse siempre, como primer recurso.
Si está dieta fracasa hay que intentar la siguiente
- Dieta oral + suplementos dietoterápicos
- Módulos grasos (MCT) e hidrocarbonados.
Evitar interferencia con la alimentación normal (después de las comidas, al acostarse).
Si está dieta fracasa hay que intentar la siguiente
- Dieta oral + Nutrición enteral
- La NE puede ser intermitente (nocturna) o continua.
Con fórmulas poliméricas hipercalóricas. Eventualmente inmunomoduladoras
Si está dieta fracasa hay que intentar la siguiente
- Nutrición Parenteral
- De corta duración, en malnutrición grave o situación comprometida (trasplante, infección grave)
El trasplante pulmonar bilateral o cardiopulmonar es el tratamiento de elección en aquellos pacientes con FQ en los que no cabe otra alternativa terapéutica Es difícil en algunos casos indicar cual es el momento adecuado para realizar el trasplante.
Aumenta la supervivencia y mejora la calidad de vida en pacientes con enfermedad pulmonar terminal. En la actualidad, la tasa de supervivencia a un año se estima entre 80%-90% y a 5 años entre 60%-70%.Los pacientes que deben ser llevados a trasplante son aquellos con expectativa de vida <50% a 2años. Para fibrosis quística, los parámetros incluyen: VEF1 <30%, hipoxia y/o hipercapnia, deterioro clínico rápidamente progresivo o hemoptisis importante. Muchos centros excluyen pacientes con infección por Burkholderia cepacia. Sin embargo, han sido descritos por lo menos 10 tipos diferentes de esta cepa y sólo uno de ellos (gemonovar III) se asocia con complicaciones y mal pronóstico luego del trasplante.
En el pasado, se consideraba como contraindicación la infección por P. aeruginosa multirresistente. Se ha demostrado que pacientes con la infección tienen pronóstico similar a aquellos con infección bacteriana sensible a antibióticos comunes.Tampoco se ha visto correlación alguna entre la colonización pre-trasplante por Aspergillus y enfermedad invasiva post-trasplante.La fibrosis hepática con hipertensión portal es una contraindicación para trasplante aislado de pulmón y en estos pacientes debe considerarse la posibilidad de realizar un trasplante pulmón hígado.
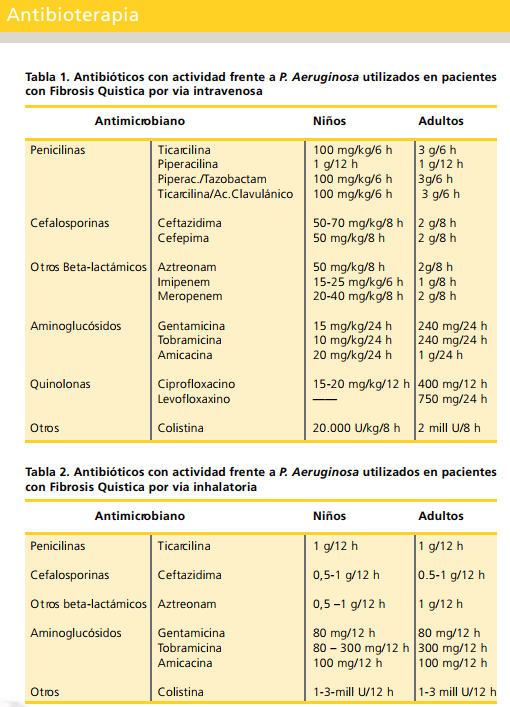
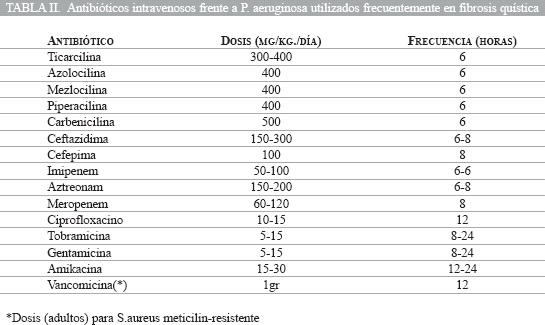
Durante las exacerbaciones se plantea el ingreso para tratar por vía intravenosa el cuadro.
Muchos pacientes presentan un cierto componente de broncospasmo, por lo que es necesario el uso beta-2-agonistas o bromuro de ipatropio.
El ADN liberado por los leucocitos destruidos en el foco inflamatorio constituye un factor determinante en la viscosidad del moco. La administración por vía inhalatoria de Dnasa recombinante ha demostrado que fluidifica las secreciones y favorece su eliminación. Otros mucolíticos, como suero salino hipertónico pueden ser eficaces.
En ocasiones es necesario el uso de corticoides para tratar la Aspergillosis broncopulmonar alérgica (ABPA) que se asocia con frecuencia a esta enfermedad o para tratar el broncospasmo que no cede con otros tratamientos.
En los casos en los que encuentre Aspergillus fumigatus colonizando la vía aérea en espera de trasplante, se utiliza Itraconazol por vía oral. También se utiliza en los casos de la ABPA asociado a corticoides.
En los pacientes con insuficiencia respiratoria el uso de oxígeno nocturno o continuo mejora su calidad de vida y les permite retrasar el desarrollo de insuficiencia cardiaca derecha y moverse.